Ferrer comunica los resultados principales del ensayo clínico de fase III ADORE en ELA

Posible diana terapéutica para la Esclerosis Lateral Amiotrófica
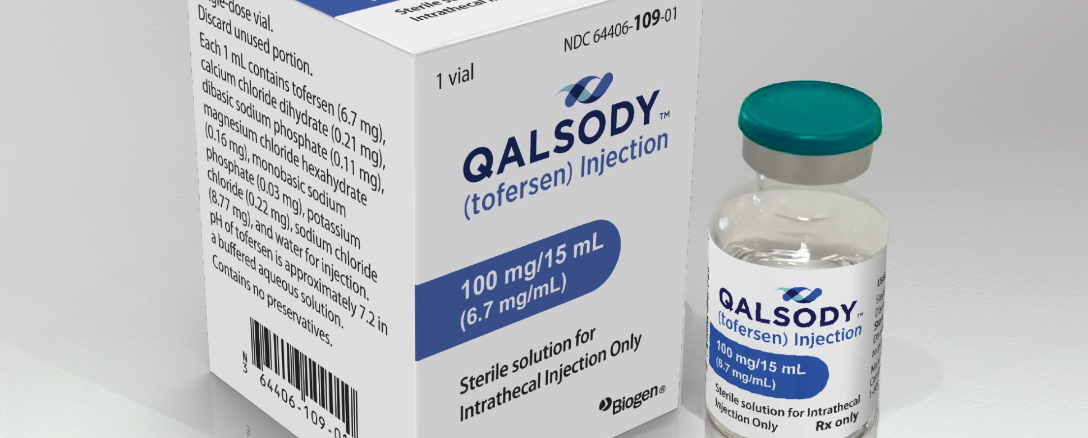
La FDA aprueba Qalsody para pacientes de ELA con mutaciones en el gen SOD1

Nueva Cátedra en Elche sobre la Esclerosis Lateral Amiotrófica

Se abre la puerta a nuevos tratamientos contra la ELA gracias a la reprogramación celular
Científicos descubren un mecanismo molecular asociado a la ELA

Primera infusión intratecal de Jacifusen en España a una paciente con ELA

Alianza europea para acelerar la investigación de enfermedades raras

Nanomateriales para el tratamiento de la ELA

Primeros pasos para autorizar un nuevo fármaco para la ELA genética

Identificados dos biomarcadores en pacientes con ELA